
Une équipe du laboratoire Généthon a mis en évidence un mécanisme jusque-là négligé dans la dystrophie musculaire de Duchenne (DMD) : le dysfonctionnement des lysosomes, que la thérapie génique par microdystrophine ne corrige que très partiellement. L’équipe a révélé sur un modèle animal de la maladie que la combinaison avec une molécule protectrice des lysosomes, le tréhalose, restaure cette fonction et améliore notablement la structure et la fonction musculaire. L’étude a été publiée dans Sciences Advances le 22 octobre 2025. Les chercheurs évryens ouvrent ainsi la voie à des stratégies thérapeutiques combinées, capables d’optimiser l’efficacité des traitements.
La myopathie de Duchenne est une maladie génétique récessive liée au chromosome X, causée par l’absence de dystrophine, une protéine essentielle à la stabilité des fibres musculaires. Son absence entraîne la désorganisation du complexe glycoprotéique associé à la dystrophine, provoquant une cascade pathologique : déséquilibre calcique, stress oxydatif, dysfonction mitochondriale, inflammation chronique et fibrose musculaire.
La thérapie génique actuelle, fondée sur l’expression d’une microdystrophine, forme raccourcie de la dystrophine, vise à restaurer une version fonctionnelle de cette protéine. Cependant, son efficacité reste partielle : les contraintes de taille qui s’appliquent sur le vecteur viral portant le gène thérapeutique empêchent l’intégration de certains domaines fonctionnels de la dystrophine native, limitant la restauration complète de la fonction musculaire.
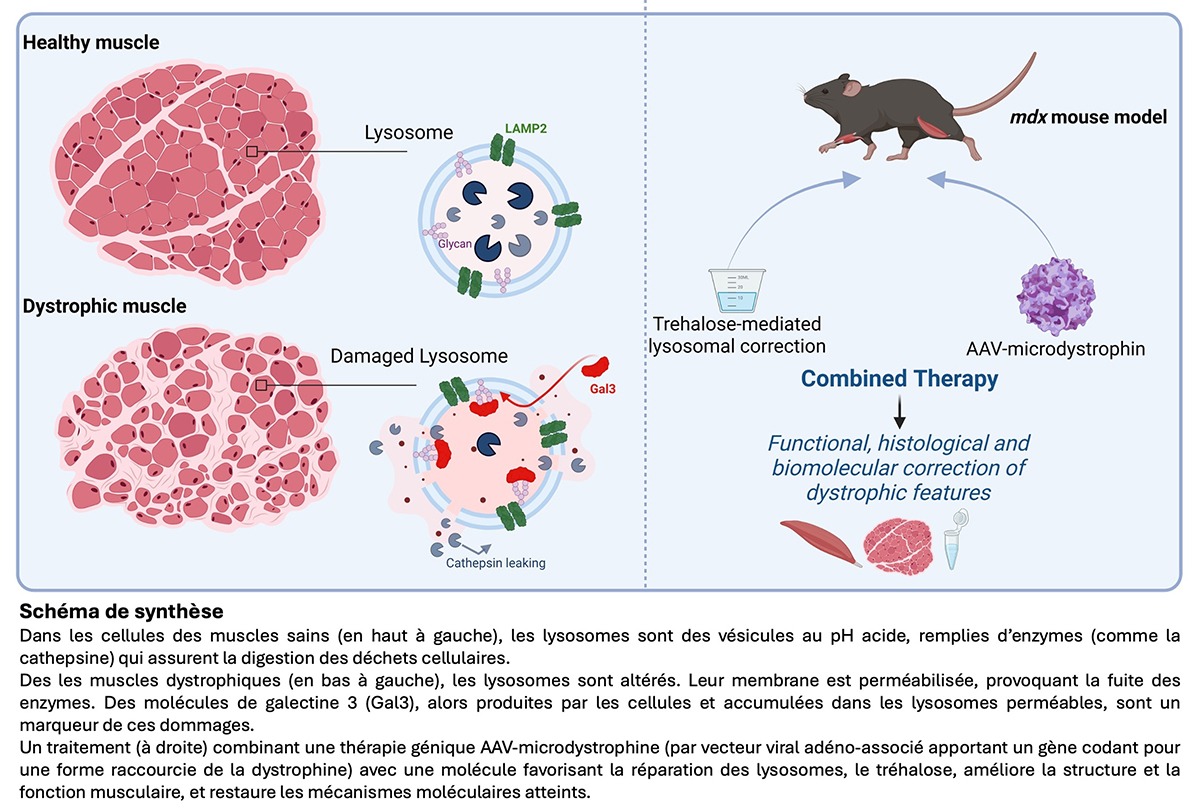
Parallèlement, des travaux récents ont mis en évidence que la myopathie de Duchenne ne se limite pas à une fragilisation mécanique des fibres : elle s’accompagne de perturbations métaboliques, notamment une accumulation anormale de cholestérol dans les muscles, liée à un dysfonctionnement des lysosomes. Présents dans toutes nos cellules, les lysosomes sont des vésicules qui renferment, dans une membrane, un milieu acide riche en enzymes. Ces organites sont essentiels à la dégradation et au recyclage des déchets issus du fonctionnement cellulaire.
Les lysosomes, une cible majeure du dysfonctionnement cellulaire dans la myopathie de Duchenne
Le chercheur Abbass Jaber, sous la supervision de David Israeli, au sein de l’équipe « Dystrophies musculaires progressives » de Généthon, a exploré le lien entre l’accumulation de cholestérol et la perturbation des lysosomes, notamment la perméabilisation de la membrane lysosomale dans le muscle dystrophique.
(Une partie de ces travaux avait été présentée par Abbass Jaber en 2024, lors de la 3e édition de la conférence « Thérapies innovantes et combinatoires », co-organisée par Genopole, l’Université Évry paris-Saclay et le laboratoire LBEPS.)
Dans des myotubes humains sains, la galectine-3, utilisée ici comme marqueur du dommage lysosomal, est faiblement exprimée. Chez les souris Dmdmdx-4Cv, modèle murin de la myopathie de Duchenne, la galectine-3 est fortement surexprimée dans les muscles et le sérum. Elle s’accumule dans les fibres musculaires en ponctuations colocalisées avec la protéine lysosomale LAMP2, signe de lysosomes hypertrophiés et perméabilisés. Les chercheurs ont fait des observations similaires dans les biopsies humaines et canines d’individus atteints.
Les analyses structurales des muscles dystrophiques ont révélé une augmentation du nombre et de la taille des lysosomes, souvent densifiés et chargés de matériel non dégradé. Les tests fonctionnels ont confirmé que l’activité protéolytique des lysosomes, mécanisme naturel de dégradation nécessaire à l’équilibre et à la survie cellulaire, est diminuée. Ces éléments démontrent une altération de l’intégrité des lysosomes caractérisée par la perméabilisation de la membrane lysosomale et un déficit de dégradation cellulaire.
Les auteurs ont montré ensuite que cette altération n’est ni transitoire ni secondaire à l’inflammation, mais qu’elle s’installe durablement dans le temps. Des résultats similaires ont été observés dans une autre myopathie, la γ-sarcoglycanopathie ou LGMDR5, indiquant que la perméabilisation membranaire des lysosomes pourrait constituer un mécanisme pathologique commun à plusieurs dystrophies musculaires.
Les expériences menées par l’équipe de Généthon sur des myoblastes (précurseurs des cellules musculaires) dystrophiques ont confirmé un défaut intrinsèque de la fonction lysosomale : après induction d’une lésion, les cellules saines réparent rapidement leurs lysosomes et restaurent leur acidité, contrairement aux cellules dystrophiques qui ne parviennent pas à activer complètement les mécanismes de réparation. Bien que des mécanismes compensatoires tels que la biogenèse lysosomale soient activés, ils ne suffisent pas à restaurer la fonction des lysosomes, comme en témoigne la persistance d’une perméabilisation de leur membrane.
Ces observations placent la dysfonction lysosomale au cœur de la physiopathologie de la myopathie de Duchenne.
Métabolisme lipidique et dysfonction lysosomale : l’engrenage fatidique
Les analyses métabolomiques montrent que la myopathie de Duchenne s’accompagne d’une accumulation anormale de cholestérol libre, d’esters de cholestérol et de sphingomyéline, résultant d’une dérégulation des gènes clés du métabolisme lipidique.
Chez les souris dystrophiques, un régime riche en cholestérol aggrave la situation. L’équipe a en effet démontré que l’accumulation lipidique amplifie la fibrose musculaire et l’altération lysosomale : le processus de dégradation cellulaire est perturbé de plus belle ; le cholestérol s’accumule à l’intérieur même des lysosomes et aggrave progressivement la dégénérescence musculaire.
Un cercle vicieux pathologique s’installe ainsi dans la pathologie de Duchenne.
Combiner thérapie génique et restauration lysosomale
La thérapie génique par microdystrophine améliore partiellement les anomalies lysosomales : elle réduit modérément la galectine-3 mais sans corriger la perméabilisation de la membrane des lysosomes. En revanche, dans la γ-sarcoglycanopathie, où la protéine complète est restaurée, le dysfonctionnement lysosomal disparaît. Cela suggère que la microdystrophine, amputée d’un domaine en tige, ne corrige pas entièrement les altérations lysosomales.
Pour pallier cette limite, les chercheurs ont testé chez les souris modèles de la myopathie de Duchenne une thérapie combinée qui associe une thérapie génique administrée à une dose suboptimale, au tréhalose, un disaccharide approuvé par la FDA qui favorise la fonction lysosomale.
L’expérience montre que le tréhalose amplifie les effets de la thérapie génique, en restaurant presque complètement la force musculaire, en réduisant la fatigue et en rétablissant la contractilité des fibres musculaires, testée ex vivo.
L’analyse histologique révèle une synergie thérapeutique : le tréhalose et la thérapie génique réduisent séparément la fibrose et l’inflammation, mais leur combinaison restaure la structure des fibres musculaires. Sur le plan cellulaire, le tréhalose réduit plus efficacement les lésions lysosomales que la thérapie génique seule, mais la combinaison des deux aboutit à un résultat comparable aux témoins sains : elle normalise la taille et le nombre des lysosomes, restaure le processus de dégradation cellulaire et de biogenèse lysosomale.
L’analyse transcriptomique confirme cette synergie : tandis que la thérapie génique par microdystrophine corrige environ 60 % des gènes dérégulés et que le tréhalose seul a un effet limité, la thérapie combinatoire restaure près de 75 % du profil d’expression. Elle normalise ainsi les voies clés comme celle de l’inflammation ou du métabolisme lipidique.
Ces résultats illustrent le potentiel thérapeutique des approches combinées dans la myopathie de Duchenne.
Vers une nouvelle génération de traitements combinatoires
Les travaux de l’équipe de Généthon ont mis en évidence le rôle central du dysfonctionnement des lysosomes dans la myopathie de Duchenne. Ils ont démontré que la correction partielle obtenue par la thérapie génique peut être considérablement améliorée par un traitement combiné ciblant la fonction lysosomale.
Au-delà de la myopathie de Duchenne, ces résultats plaident pour une médecine combinatoire pour des maladies génétiques rares, mais également des maladies fréquentes pour lesquelles un traitement unique ne suffit pas à corriger l’ensemble des désordres cellulaires.
Souvent, les pathologies sont complexes, altérant une fonction ou un organe principal mais entraînant d’autres dysfonctionnements, parfois multiples, voire en « effet boule de neige » comme illustré ici.















