L’équipe génopolitaine SysFate, dirigée par Marco Mendoza, a conçu une technologie brevetée capable de cartographier, sur des coupes de tissus déposées sur des puces à ADN, des signatures moléculaires de la régulation génique.
Fondée sur l’analyse des modifications d’histones, cette méthode d’épigénomique spatiale, la 2e conçue au monde, se distingue par son faible coût et sa capacité à analyser de grandes surfaces de tissus.
Validée sur des modèles complexes, notamment des tissus osseux fixés, elle ouvre une nouvelle voie d’exploration de la régulation, en particulier pour les tissus réfractaires à l’analyse transcriptomique, avec des applications prometteuses en recherche biomédicale et pathologique.
Explorer l’activité des tissus biologiques est le point de mire des scientifiques qui cherchent à comprendre les mécanismes biologiques, à élucider comment les pathologies les perturbent, à identifier ainsi les cibles moléculaires impliquées pour accélérer la recherche de traitements. Bien que les récents développements « omiques » à résolution spatiale soient en passe de révolutionner l’exploration de la complexité tissulaire, les solutions technologiques livrent difficilement une image instantanée de l’activité des tissus.
La plupart se concentre en effet sur la détection de la présence d’ARN, première étape du phénomène de transcription-traduction des gènes en protéines, en situation d’activité cellulaire. Or, la littérature scientifique indique que la demi-vie des molécules d’ARN dans la cellule varie de 40 minutes à 9 heures, notamment selon la fonction de la cellule (Tani et al. 2012). Ces données suggèrent qu’un profil transcriptomique n’est pas le reflet instantané de l’état transcriptionnel du tissu au moment de la mesure, mais le témoin de son activité sur un laps de temps qui peut être bien plus large. De plus, par définition, une analyse transcriptomique n’identifie que les gènes exprimés : elle ne peut donc pas distinguer, parmi les gènes transcriptionnellement inactifs, les gènes réprimés des gènes momentanément passifs mais maintenus dans un état transcriptionnel favorable.
Pour résoudre ces questions, l’équipe Atige* SysFate (cf. photo), créée et dirigée par Marco Mendoza au sein de l’Unité de Génomique métabolique (Genoscope – CEA / CNRS / Université d’Évry), a mis à profit sa double expertise en omiques et en bio-informatique pour développer une technologie de cartographie qui capture l’activité transcriptionnelle à l’instant « t » sur chaque point de sections transversales de tissus, à partir de l’analyse de la chromatine. La méthode a fait l’objet d’un brevet et d’une publication en juin 2025 dans Genome Research.
La chromatine porte la signature de l’état d’activité transcriptionnelle
Dans le noyau des cellules, l’ADN n’est pas libre mais enroulé et compacté autour de protéines particulières appelées « histones ». L’ensemble forme la chromatine.
Les modifications post-traductionnelles des histones constituent un système dynamique qui joue un rôle essentiel dans la régulation de l’expression des gènes. Ces modifications incluent notamment des méthylations, acétylations, acylations d’acides aminés spécifiques de ces protéines. Elles déterminent l’état des gènes auxquels ces histones sont associées, actifs, réprimés, ou encore bivalents, c’est-à-dire non exprimés mais facilement activables. Les modifications d’histones font partie des mécanismes dits « épigénétiques » qui ne modifient pas la séquence d’ADN mais constituent des marques moléculaires associées physiquement aux chromosomes et capables de se transmettre au cours des divisions cellulaires. Elles interviennent notamment au cours du développement embryonnaire, lors de la différenciation cellulaire, ou encore dans des phénomènes pathologiques comme les cancers.
Capturer les modifications d’histones, clés de voute de la régulation épigénétique
Aujourd’hui, la stratégie d’immunoprécipitation de la chromatine, c’est-à-dire l’utilisation d’anticorps pour capturer ces protéines associées à l’ADN et caractériser des profils d’interactions entre ADN et histones, est devenue la méthode de référence pour interroger l’état de régulation de la transcription au moment « t » sur des prélèvements de tissus (Bannister et Kouzarides, 2011). Initialement, cette approche reposait sur une fragmentation mécanique de la chromatine (sonication), mais des modifications récentes ont décliné une version enzymatique de la technologie (« Cut&Tag » ; Kaya-Okur et al., 2019), de grande spécificité et de haute sensibilité. Elle réduit le nombre de cellules requises de millions à quelques milliers, compatibles avec l’analyse de petits tissus, voire l’échelle de la cellule unique (Bartosovic et al., 2021).
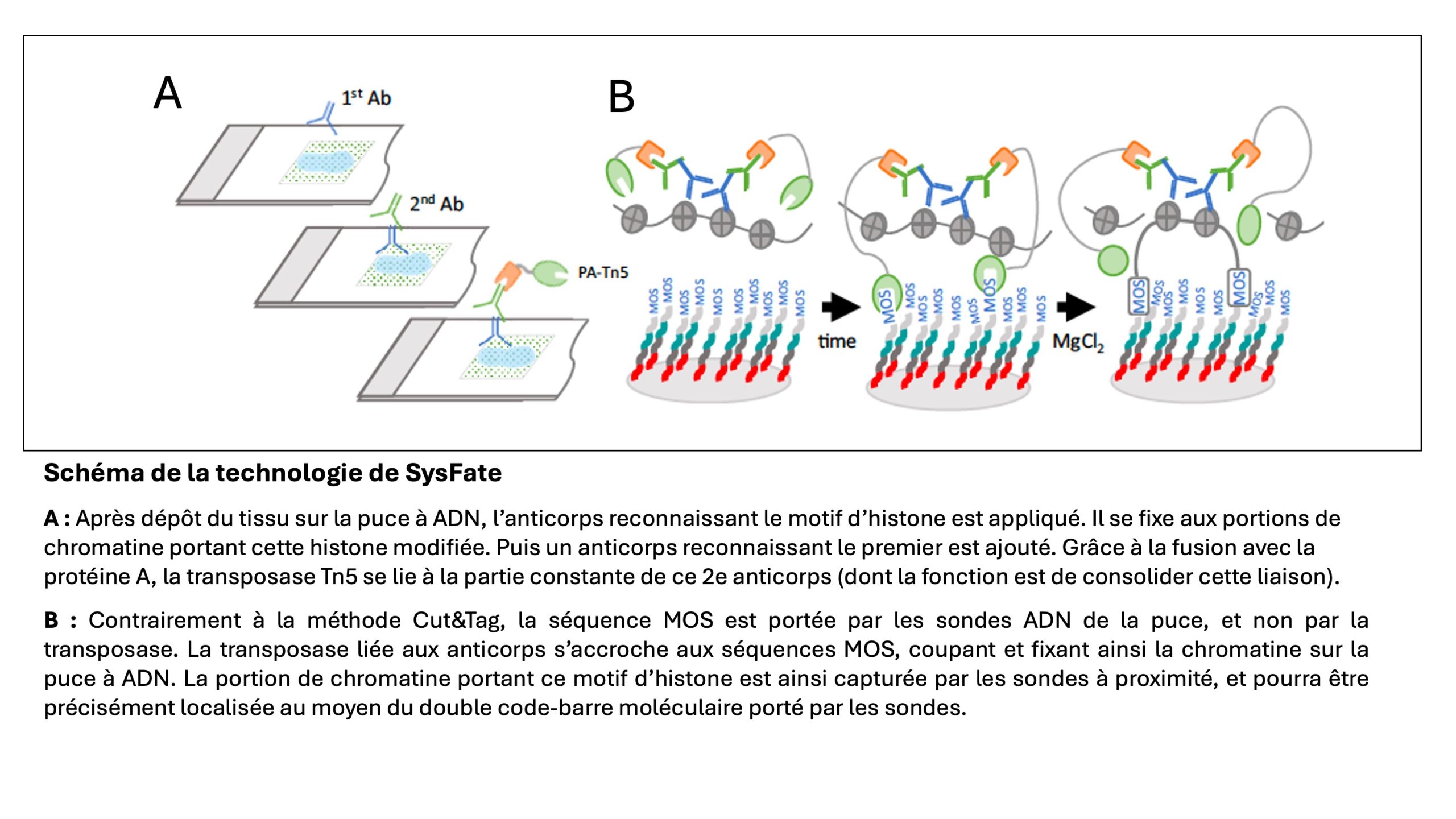
La méthode Cut&Tag utilise :
- (1) des anticorps ciblant les différentes modifications d’histones,
- (2) une protéine bactérienne (protéine A) de forte affinité pour les régions constantes des anticorps et
- (3) une transposase modifiée (Tn5), fusionnée à cette protéine : les anticorps se fixent spécifiquement aux motifs des histones puis grâce à la fusion protéique, la transposase se lie aux anticorps fixés sur la chromatine. Elle coupe l’ADN à proximité et insère, via de courtes séquences d’ADN dites « MOS », des adaptateurs qui vont permettre le séquençage du fragment ainsi constitué.
Il est ainsi possible de détecter des modifications spécifiques d’histones associées à l’état de régulation de l’expression de gènes (méthylations, acétylations… d’un acide aminé à une position donnée de l’histone) et d’identifier les gènes en question.
La technologie d’épigénomique spatiale de SysFate
L’équipe SysFate a développé et breveté une nouvelle technologie, adaptée de la méthode Cut&Tag, qui, grâce à une puce à ADN, capture et localise sur des coupes de tissus ces signatures de modifications d’histones, avec une résolution de 100 micromètres.
L’innovation repose sur la conception de cette puce à ADN sur laquelle le tissu biologique, préalablement perméabilisé pour donner accès à la chromatine, est déposé, et sur les étapes de biologie moléculaire pour recueillir l’information captée par la puce.
Celle-ci porte d’une part, un double code-barre moléculaire ligne-colonne (en savoir plus) pour localiser précisément chaque point analysé et d’autre part, la séquence MOS pour que la transposase s’y accroche, puis coupe et fixe la chromatine à cet endroit (cf. schéma) : en immobilisant le tissu biologique et plus précisément le fragment de chromatine portant le motif ciblé de l’histone, la méthode mise au point par SysFate produit une image instantanée, en 2 dimensions, de l’état de régulation dans le tissu.
Explorer les tissus complexes et les échantillons récalcitrants à la transcriptomique
Marco Mendoza et son équipe ont validé cette méthodologie sur des cerveaux de souris adulte, sur des embryons de souris, ainsi que sur du matériel biologique osseux préalablement décalcifié et conservé dans des blocs de paraffine (FFPE).
Dans ce dernier cas, il s’agissait d’étudier un modèle murin de polyarthrite rhumatoïde, et notamment d’examiner les mécanismes de régulation au niveau de l’articulation. Les analyses du tissu osseux nécessitent une décalcification et une conservation en paraffine qui détruisent en partie l’ARN, rendant difficile l’application des méthodes de transcriptomique.
Peut-on exploiter de tels tissus avec la technologie d’épigénomique spatiale ? c’est la question à laquelle SysFate a répondu positivement au cours d’une thèse Cifre menée avec Novalix.
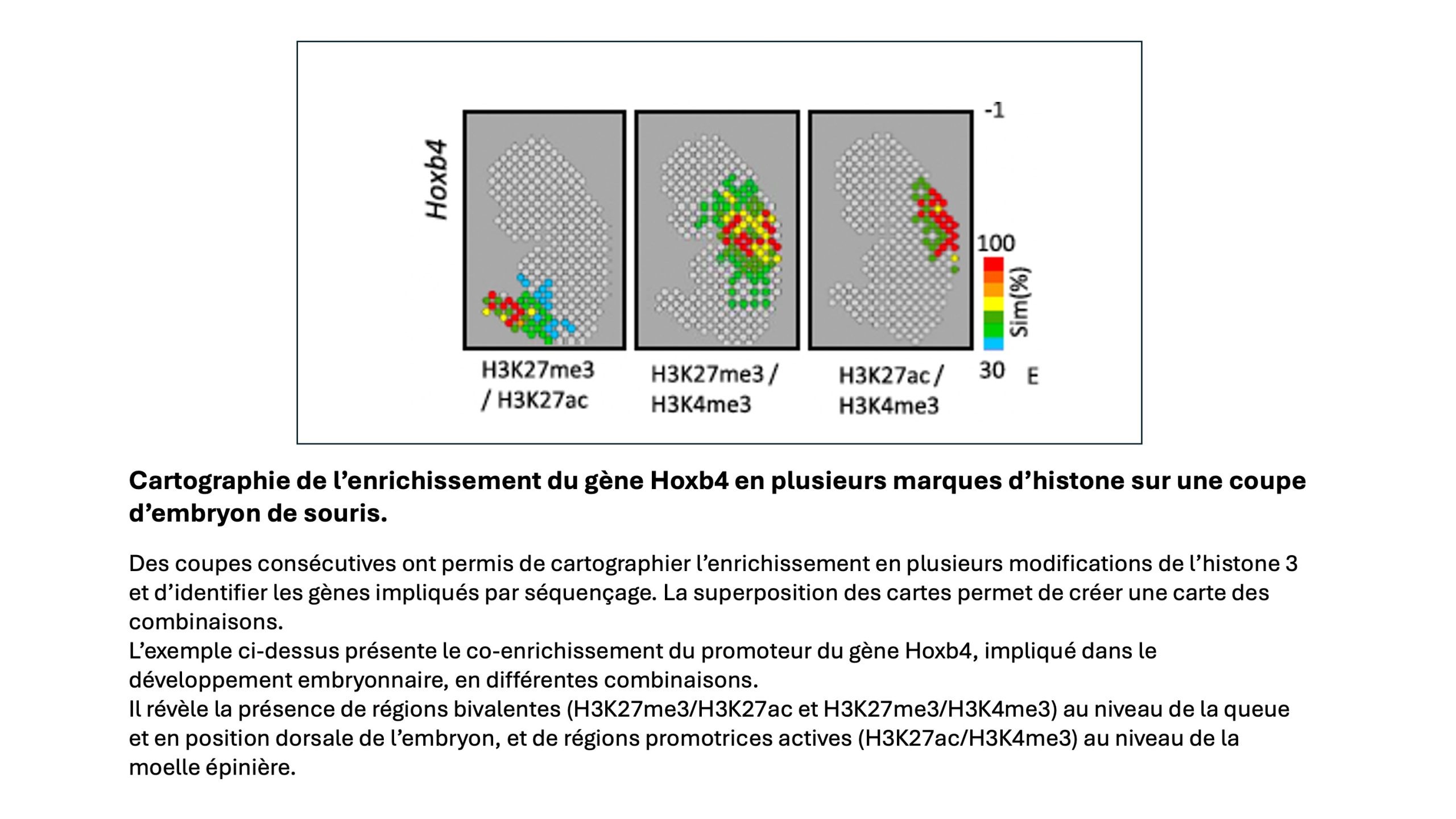
Dans les pattes de souris modèles de la polyarthrite rhumatoïde, des gènes comme Itga2 ont révélé un enrichissement en H3K27ac (acétylation de l’acide aminé lysine en position N-terminale 27 de la protéine histone H3), qui signale une activation de la transcription. L’équipe a appliqué sa technologie à des coupes sagittales de pattes de souris, issues du protocole de décalcification-fixation, déposées sur une puce à ADN de 2048 sondes. Les chercheurs ont obtenu une carte des positions sur le tissu où la chromatine est enrichie en H3K27ac. Le séquençage leur a permis d’identifier, par position, environ 1800 gènes concernés, plus exactement des promoteurs de gènes, intervenant notamment dans la fonction ostéocytaire (Pdpn), dans l’ostéoblastogenèse (Dlx5), associés aux cytokines pro-inflammatoires (Il1b, Il6, Il10) ou encore à des récepteurs connus pour être surexprimés dans la polyarthrite rhumatoïde (Gabbr1).
cf. ci-dessous : cartes d’enrichissement en modification H3K27ac des promoteurs de 12 gènes

L’équipe a comparé les résultats obtenus à des données publiques provenant soit d’analyses d’interactions ADN-protéine par ChIP-seq (immunoprécipitation/séquençage), soit de la recherche des transcrits par RNA-seq. Toutes sont issues de la base de données génomiques qualifiées, conçue par Marco Mendoza en 2019. L’analyse comparée confirme la corrélation entre l’enrichissement en H3K27ac révélé en épigénomique spatiale et le niveau d’expression génique.